{kind=link}
Here's something you might be interested in.
Ask a Hipster — Advice you didn't know you needed
Big Screen — Movie commentary
Blurt — Music's inside track
Booze News — San Diego spirits
Classical Music — Immortal beauty
Classifieds — Free and easy
Cover Stories — Front-page features
Drinks All Around — Bartenders' drink recipes
Excerpts — Literary and spiritual excerpts
Feast! — Food & drink reviews
Feature Stories — Local news & stories
Fishing Report — What’s getting hooked from ship and shore
From the Archives — Spotlight on the past
Golden Dreams — Talk of the town
The Gonzo Report — Making the musical scene, or at least reporting from it
Letters — Our inbox
Movies@Home — Local movie buffs share favorites
Movie Reviews — Our critics' picks and pans
Musician Interviews — Up close with local artists
Neighborhood News from Stringers — Hyperlocal news
News Ticker — News & politics
Obermeyer — San Diego politics illustrated
Outdoors — Weekly changes in flora and fauna
Overheard in San Diego — Eavesdropping illustrated
Poetry — The old and the new
Reader Travel — Travel section built by travelers
Reading — The hunt for intellectuals
Roam-O-Rama — SoCal's best hiking/biking trails
San Diego Beer — Inside San Diego suds
SD on the QT — Almost factual news
Sheep and Goats — Places of worship
Special Issues — The best of
Street Style — San Diego streets have style
Surf Diego — Real stories from those braving the waves
Theater — On stage in San Diego this week
Tin Fork — Silver spoon alternative
Under the Radar — Matt Potter's undercover work
Unforgettable — Long-ago San Diego
Unreal Estate — San Diego's priciest pads
Your Week — Daily event picks
San Diego cystic fibrosis sufferers learn to live life by the moment
No time to dream

Intensive chest therapy involves physically beating on the patient’s chest.
"When I was nine my grandmother heard from a friend about this clinic down in Mississippi on the Gulf Coast. So my mother and I flew down there in the winter because I was constantly up and down, sick some days, well the next — missed a lot of school. We went down to this miracle clinic where they gave you this elixir with arsenic in it; stayed there a week, must have been a very expensive thing. We came back and I started taking the miracle elixir; did that a few months and my parents noticed I was going bald — I was losing all my hair. So they stopped the elixir and miraculously my hair started growing back... I don't think it changed things much, the miracle elixir didn't do a whole lot, but it tasted horrible, I do remember that. It was pink and it tasted like the inside of somebody's tennis shoe. When I had to take that during the day it was like, oh no!! But I took it because I thought I was going to get better; none of us knew about CF. We were ignorant. We were babes in the woods." — Bob Bourquin

Bob Bourquin. When Bob walks in, is 26, looks like a football player, those parents have got to feel some resentment.
Ellen Derner is 29 and works in a southeast San Diego kindergarten as a teacher’s aide. Her father shied away from the miracle elixirs, but 20 years ago, when she too was nine, he quit his job, sold everything that wouldn’t fit in the back of a U-Haul, loaded Ellen, her mother, and two sisters in the family car, left his native Montreal, and started driving south — to somewhere it didn’t snow. Ellen had spent the previous summer with an aunt in Florida, where the series of colds and the terrible, persistent cough which had prompted the desperate move seemed to have subsided. Florida was too humid though, so they drove west. When they reached Arizona, Ellen stopped coughing, but her mother didn’t want to live in Arizona and they continued on to Los Angeles. The smog in L.A. was unbearable so they again turned south, coming to a stop in San Diego.

Ellen Derner: “As it turns out, the climate doesn’t make any difference."
“As it turns out, though,” says Ellen, “the climate doesn’t make any difference, and about a year after we got here I got sick again. It was diagnosed [falsely] as tuberculosis and I was bedridden for eight or nine months…they were pretty sure I was going to die. My parents regretted we’d left Canada then.” But they stayed, suffering through the unexplained illnesses and colds until a firm diagnosis was made when she was 19.

Mark Longfellow.was eating his mother out of house and home — 18 pancakes at one sitting.
At that time she went to University Hospital, where a test was performed in which a weak electrical current and a special chemical were applied to her arm, causing a small area of skin to sweat. After careful collection and measurement, it was determined that her sweat contained an abnormally high amount of salt; the test was positive. However, her insurance plan required that she go to another hospital for treatment, and there she was retested three times, each time with a negative result. However, she was somewhat dubious about their technique. “They put my whole body in a plastic bag with just my head sticking out the top and waited for me to sweat so they could collect and measure it.” But it could have been worse, according to Ellen. “They used to wrap kids in wool blankets and make them run around the hospital.”

Rita Bowers: "No, I’m not ready to die. I’ve still got too much to do."
She herself never doubted the outcome of the tests. “I knew I had all the symptoms. So I finally went back to University and paid to have it redone.” Positive again. She had cystic fibrosis.
The disease first appeared in the scientific literature in 1936 as congenital cystic pancreatic fibromatosis and bronchiectasis syndrome, a mouthful of a name that basically meant doctors were finding some very sick babies with a puzzling combination of symptoms previously thought unrelated: chronic lung infection in patients who also developed fibrous and cystlike scarring of the pancreas.

Dr. Ivan Harwood “can’t tell anybody they’re going to get better."
By 1938 researchers were satisfied that the strange combination was caused by a single disease and they trimmed the name to fibrocystic disease of the pancreas, but still not a lot was known about it. Doctors were telling mothers, “We don’t know why it is, but these babies all follow the same pattern. You can’t find any food to agree with them, and then they get pneumonia and die.” When the accepted name became cystic fibrosis a few years later, the disease had become distinguished, said one textbook, “for the frequency with which it was correctly diagnosed for the first time at post mortem.”
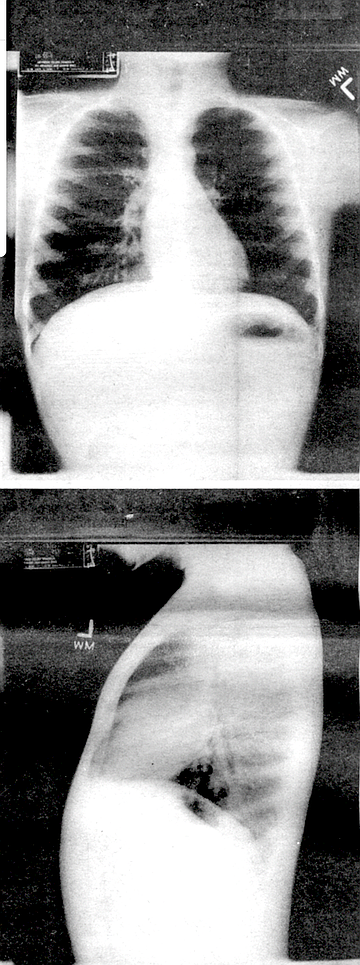
CF patients cannot properly expel the thick secretions they produce and the mucus remains trapped in the lungs.
A more useful method of diagnosis was developed as a result of observations made during a New York City heat wave in 1948. As is common during a heat wave, hospital emergency rooms began filling up with victims of heat stroke brought on by excessive salt and fluid loss. A pair of doctors noted that a curiously high number of these persons seemed to have cystic fibrosis and they began wondering why. It was soon discovered that the sweat of these patients contained up to ten times the normal amount of salt — explaining the high incidence of heat stroke and also forming the basis for a diagnostic test.
Yet it is unfortunate that the sweat test Ellen Derner described is so often incorrectly done, because it is the only conclusive test for an incurable and poorly understood disease. Cystic fibrosis (CF) occurs in one-fourth of the offspring from two parents who carry the genetic trait; it is the greatest genetic killer of young people in this country, with as many as 1 out of every 20 Caucasians believed to carry the gene. Such carriers are not affected by the disease, however, and since they show absolutely no symptoms, cannot be identified; despite the highly promising work of several researchers, at the moment the only sure way of identifying a carrier of CF is by working backward: the parents of a child with CF must carry the gene.
The basic genetic defect which is responsible for cystic fibrosis remains unknown, but it is known that this defect causes excessively thick and sticky secretions of mucus. This thick mucus brings on an almost unbelievable variety of problems, the most serious being chronic and progressive lung infections, and inefficient digestion resulting from clogged digestive ducts. Mucus is produced by the lungs as their primary means of fighting infection (which is why people get congested when they have a cold), but CF patients cannot properly expel the thick secretions they produce and the mucus remains trapped in the lungs, where it fosters the growth of bacteria — which in turn promotes the secretion of more mucus, which then provides a home for more bacteria. It is this circular pattern of lung infections which eventually proves fatal.
In the ’50s a CF child could not be expected to reach school age. By the mid-’60s the average life expectancy had grown to age 11. Now, with better treatment and earlier detection, the national Cystic Fibrosis Foundation projects that half of all CF patients born today will live past the age of 21. But increased longevity has in many cases become something of a mixed blessing for longer-lived patients who have become grown-ups burdened with a disease perceived almost exclusively — both by the general medical community and the public — as an affliction of children only.
The executive director of the Cystic Fibrosis Foundation’s local chapter is Winnie Burke, an energetic and outspoken woman in her mid-40s who directs the chapter’s fund-raising and public-education activities from her office on Fourth Avenue in Hillcrest. “When I first took this job,” she says, “the first time I got close to a kid who died, I sat here in my office and I cried and cried. The other woman who was working in the office at that time came in and she said, ‘If you’re going to react like that every time one of these kids dies, I don’t think you’re going to be suited for this job.’ I looked at her and I said, ‘If I don’t react this way, I don’t think I’m going to be suited for this job.’ ” In the intervening ten years, Burke has had no shortage of opportunities to adjudge her job fitness, since this series of events repeats itself in her office with distressing regularity.
Like virtually anyone whose life has been touched by cystic fibrosis, Winnie Burke learned its lessons quickly. Doctors have called it “the great masquerader.” It can be subtle, whimsical, merciless, and above all, arbitrary. For a CF patient, life is a misshapen path of sharp turns and detours about which nothing is predictable except that it always leads downhill, sometimes gently, sometimes steeply. The disease is maddeningly erratic; what happens to one person is no indication of what will happen to another. Within a single family one child might die at a few years of age while another makes it into his 30s to become one of CF’s “old folks.” But such reprieves are understood to be only temporary. Today’s apparent good health, the brisk seven-mile run, the 75-mile bike ride, the workout at the gym, all too frequently give way to next week’s ten-day hospital stay. One minute’s laughter becomes the next minute’s wrenching cough, which, sometimes, leads to the burst artery and coughed-up blood of hemoptysis. And if it doesn’t, there is always the fear.
If these are extremes — the more common story of CF being one of frail, barrel-chested children, of frequently missed school and canceled family vacations — they are not atypical; it is a disease which defies neat description. It is a disease Camus or Sartre would have understood, for it teaches the meaning of existentialism as few things can. It is a disease which causes five- and six-year-olds to think deeply about sorrow and joy, the unfairness of fate, and why people are born to die.
In 1967 cystic fibrosis became part of the life of an attractive, composed Coronado woman named Judy Longfellow. Her son, Mark, was 14 months old at the time and she recognized, she says, “that something was wrong. He had diarrhea an awful lot and was eating me out of house and home — 18 pancakes at one sitting, that sort of thing; it was incredible. It was some time before we came up with the diagnosis, because after we had tried eliminating different foods [to check for allergies], we did a sweat test at Mercy Hospital which came back negative. But our doctor was very suspicious that Mark had CF, so we took him up to Children’s Hospital for more work. They did several tests and they all came back positive.
“There was a panel of doctors, there were like three, and we went into this room — Ralph [her husband] wasn’t with me. It was a very dark dismal kind of room, a room that would hold maybe 30 people, and we sat in this little nucleus and they basically told me Mark had CF, and they didn’t really elaborate much at all except to say that it was a very harsh disease. Then they sent me on my way.”
But a few days later Mrs. Longfellow and her husband were provided with a few details by their specialist at Children’s.
“He came into the room and he said, ‘Well, you might as well know right now that if your son coughs, that’s just about the end; he will probably not last three months. He’ll never be able to go to school.’ The picture he painted for us was very, very black. So we were left with absolutely no hope that this child would live to be two years old.… His manner was very matter-of-fact, he was in the room maybe three minutes; he went through all this stuff and then he was gone. That angered me considerably.”
Mark first exhibited the deep, thick cough of CF when he got a lung infection about six months after this. The doctor’s three-month time limit proved wrong, however, and Mark did go to school — where he excelled. He was tested at the age of six and found capable of doing college-level math. “He loved math,” said his mother, “just loved it.” He was also an avid baseball player and one year he became a Little League all-star. Not until last spring, when Mark was 14 years old, did he die.
Mark was a child with a lot of friends, and in spite of his disease he led an active life, but it was a life punctuated by visits to the hospital. In contrast to some CF kids, who by the age of 13 may have been in as many as 30 times, Mark, says his mother, “wasn’t in really a lot. Maybe seven times. There was one period where he went for almost two years without a hospital stay.”
Usually such hospitalizations are for what is referred to in CF jargon as a “tune-up.” Tune-ups become necessary when a cold stirs up the bacteria in a CF patient’s lungs; they usually last 10 to 14 days, and their main treatments are the administration of intravenous drugs and intensive chest physical therapy that involves physically beating on the patient’s chest. This can be done with either a mechanical vibrator or by hand in a clapping manner which evokes the sound of a horse’s clippity-clop; a few minutes spent observing this procedure can yield a keen perspective on the limitations of modern medicine. The purpose of this activity is to loosen and dislodge the thick, sticky mucus (termed “junk”) which a person with CF can cough up in mind-boggling quantities after a treatment. One adult commented that he coughed up eight ounces a day when he felt well and twice that when he had an infection.
CF patients are advised to do treatments at home as well as in the hospital; they can be done by a child’s parents, or, when old enough to use the special vibrators, children can do their own. The Longfellows came to view the treatments as a part of life, a habit “like brushing your teeth every morning. It took an hour every time we did them and we routinely did them twice a day.” But while some patients have no doubt about the value of treatments (one proclaiming, for instance, that “if I stopped doing my treatments today I would probably be in the hospital in two weeks, maybe not even that long”), others avoid them fanatically because they symbolize the unremitting grip CF maintains upon those it affects: two, three, sometimes up to four hours a day spent clearing one’s lungs in a ceaseless effort simply to stay alive. One mother said, “It’s just like pulling hen’s teeth” to get her 11-year-old son to take a treatment. “He hates it. He thinks if he works on himself he doesn’t need that pounding. In the morning he goes in and he coughs and he coughs and he gets most of that stuff up himself. He’ll cough for maybe five or ten minutes.”
It is one of CF’s more unnecessary tragedies that the type of callous (and often ignorant) welcome to the medical world which Judy Longfellow received is far from uncommon. And though families have had the amazing luck of having a child correctly diagnosed the very first time he got sick, the stories of wrong diagnoses, missed diagnoses, and refused diagnoses are legion among the CF community. One man told of having “pneumonia, or some problem, as an infant”; “repeated illnesses” all through his youth; “what was diagnosed as viral pneumonia three times” in his early teens; “mononucleosis during college”; “and other problems off and on.” One physician was “convinced” he had tuberculosis and “kept running skin tests. He couldn’t understand why they kept turning out negative.” He spoke of being given 40 different diagnoses — with any one as good as another — and of being treated for “allergies” until his mid-20s.
It was the ironic ill fortune of this man, now 35, to be too healthy and longlived to be considered a candidate for cystic fibrosis. Nor is this sort of episode confined to the backwoods of medicine: as recently as two years ago, and based upon the erroneous results of a sweat test conducted by placing a plastic baggie over one hand, a pulmonary specialist at one of San Diego’s most respected medical institutions persisted in treating for tuberculosis an adult patient who had been clearly diagnosed elsewhere as having CF — presumably because the patient was too old and too well to be “cystic.”
Just as common as the litany of false diagnoses and incorrect treatments are the attendant tales of mental turmoil compounded by physicians who failed either to recognize the medical subtleties and variability of CF or to comprehend and appreciate the emotional difficulties generated by the disease, or both. When Ellen Derner was finally diagnosed, she was understandably relieved, as she put it, to “find out what it was, to learn that the symptoms were all related and that it had a name.” But her doctors, she says, couldn’t understand that. “One of them stood in my room and yelled at me that this was something that was going to kill me and that I had better start taking it seriously. They couldn’t understand what a relief it was to have it all explained, to have it all make sense.”
The medical mistakes are understandable given that virtually nothing in CF is orderly — diabetes, liver disease, and sterility can develop in addition to the respiratory and digestive disorders; and it can be found among blacks and Hispanics, though it is disproportionately a disease of whites. But what is harder to understand is that the predominant attitude within the medical community seems based on an image of CF drawn largely from the ’50s. The general medical community maintains a picture of CF similar to that put forth recently by the prestigious New England Journal of Medicine. In an editorial, they pronounced CF “a grim sentence, usually prolonged through an unhappy adolescence to a sad, inevitable end.” This attitude, unfortunately, is often reflected in the medical care dispensed by physicians who hold it, and it is this situation which contributes to the unrestrained joy most CF patients and families feel when they finally do find a physician familiar with their disease.
The San Diego Cystic Fibrosis and Pediatric Pulmonary Disease Center at University Hospital is 1 of 125 specialized centers supported by the Cystic Fibrosis Foundation. The medical care of approximately 160 local CF patients is supervised here by Dr. Ivan Harwood, center director, and Dr. Nancy Olmsted, center codirector.
The center’s administrative facilities and offices are located two blocks south of the hospital in a former residence on Front Street that has since been named “CF House.” One of these offices is used by Dr. Harwood; its appearance tells a great deal about the man who, says one of his patients, “can’t tell anybody they’re going to get better. They can plateau and remain stable, but all of his patients are going to die.” On the walls hang photographs of children he has cared for; in a corner sits an old wooden desk littered with papers and a wide assortment of items: a microscope which appears older than van Leeuwenhoek’s; a small ceramic elephant; a rack of smoking pipes; and the disordered components of a unique filing system referred to by others in the office as the CFIS — the Cystic Fibrosis Information Sink, into which correspondence and other printed matter often vanish for wondrously long periods of time.
Ivan Harwood came to San Diego in 1970 and joined the faculty at UCSD a year later. He came at a time when about half a dozen patients were being seen twice monthly at a clinic established through the efforts of the local CF chapter. He didn’t come to San Diego intending to run the CF Center. “I didn’t plan it,” he says. “I sort of fell into this backward. There were a few people who needed care and there was a need that wasn’t being met.” There was also another appeal, something Harwood refers to as “the chemistry between a few patients and a doctor.”
Few physicians are attracted to the care of the chronically ill. It subverts their image as healers; to say it can be disheartening is an understatement. One member of the hospital CF team estimates that they lose roughly one patient every six weeks. This often leads doctors to immerse themselves in the scientific aspects of medicine, depersonalizing the patient and the disease. Avoiding this form of self-protection has required Dr. Harwood to develop a perspective which takes into account the limitations of specializing in a disease without a cure. “If I approached CF as a disease where I always lost or where there was nothing to be done,” he says, “it would be so dismal and hopeless that I couldn’t continue to be involved with it very long. So I think what I’ve done is to say well, okay, my role as a physician is not necessarily just to cure a disease — because I can’t cure this one, I accept that from the start — but I can do an awful lot at some stages of the disease to prolong life, and I can do certain things in the end stages to make a very difficult part of one’s journey through life a little better, a little easier. So I’ve gotten to the point where I say, well, that’s part of the whole business. It’s difficult, I accept that, but I also have the satisfaction of being competent with patients in that very difficult stage — patients that usually, because of the nature of the disease, we have gotten to know very well and to care about.”
The San Diego CF Center utilizes a “team approach” in caring for its patients. In one sense the team is the group of professionals responsible for ministering medical care in all its forms — the doctors, nurses, technicians, therapists, and social workers. But as an outgrowth of Dr. Harwood’s belief that “the ultimate responsibility for somebody’s care is in themselves or their parents,” patients and families are also strongly encouraged to take part in decisions concerning their medical care. “It really works better for us to make the family and the patient partners in the whole process of trying to manage a disease that can’t be cured.”
A dual perspective on CF and its care is offered by one respiratory therapist at University Hospital. Bob Bourquin has CF in addition to being a member of the medical team. He is 26 (and still has his hair despite his subjection to the “miracle elixir cure”); he says there are some absolutes in CF “and there’s an absolute with me: I’m going to live another five years. It’s not definite but I think that’s reasonable.”
Growing up in Maryland, he spent a sickly childhood with “asthma” and “allergies” until the day he “turned purple” when he was nine. “I couldn’t breathe at all. It was just like I was shut down.” He recovered after a prolonged hospitalization during which doctors made the diagnosis of CF and told his parents, “Your child has cystic fibrosis and he’s going to die.”
He spent a lot of time after that trying to deny he had CF. “That’s pretty much how I approached the thing when I was in my teens. Even when I was really sick I’d never do treatments. I could be so sick I could climb a flight of steps and almost black out — in school the 50-yard dash was more like the 50-yard crawl — and still I wouldn’t do anything for it.
“I knew so little about CF six or seven years ago. So, so, very little. And you have all these preconceived notions which you get from the newspaper articles. Like somewhere I’d read the average life span of somebody with CF was 21. I figured I was 19; I had two years to live. I thought that was it — everybody died when they were 21. I didn’t realize at the time, because I was totally uneducated medically, that some people live a lot longer, some people live a lot shorter. I really thought that when I turned 21 that would be like the last supper. I’d cut the birthday cake and I’d go in and lie down. And die.”
When he was 19 an incident occurred which forced him to realize what CF would mean: he spent five weeks in the hospital as a result of air leakage from his lungs into his chest cavity — a pneumothorax. “That was the big turning point. I could not deny it anymore. I started doing treatments every day. I spent the entire fall of that year like a self-imposed cripple, paranoid of having to go back in the hospital. I was extremely depressed, did nothing except lie around the house in utter paranoia and despair.”
Shortly after this Bourquin began obtaining medical care from a CF center in Cleveland. His spirits started to improve as he met other CF patients and grew to realize that he could take a direct hand in his medical care. “It’s not sheer happenstance that I’m where I am now. I work hard at it. And it is hard to get in an hour and a half, because that’s how long my treatment takes me every day. It’s hard to do that and still go to work and do everything else.”
Remembering the reassurance that can come from meeting other, especially older, CF patients, Bourquin makes it a point to spend part of his time at University Hospital talking with the parents of CF children. It is a task he has learned to approach with delicacy. “If the children are fairly healthy, or even moderately healthy, the parents are universally pleased, relieved, and delighted to see me there,” he says. “They can look at me and say, ‘Wow, that’s neat, maybe my kid is going to grow up.’ If, on the other hand, their kid’s really sick — when one of the CF kids is dying and I’m the therapist — that’s when I really get the resentment vibes, or the jealousy vibes. You know, when Bob walks in, is 26, looks like a football player, I feel somewhat guilty. Those parents have got to feel some resentment; at that point they know their kid’s not going to make it and they can’t help but be jealous that I have. That’s just basic human nature.
“There’s a whole lot of vibrations between CF patients. It’s very awkward when you’re healthier. Vibrations go out to you of jealousy and anger — I’ve been the recipient of those. When you see somebody who’s sicker than you it’s tough because your heart really goes out to them. You know yourself when you’ve been sick how you feel, and when you see someone with CF who’s dying, who’s close to death, it’s hard to look at them because you know what it’s like and you know what it’s going to be like; you just sort of are instinctive about it. On the other hand, it’s the old animal instinct: thank God it isn’t me.”
Other reactions can also mark the relationships formed between people with CF: comfort and relief can come through learning that others share the same plight, the same experiences, the same worries; despondency can result from watching friends fall like so many dominoes, from thinking, “Here are X number of young adults and Monica died, and so-and-so died, everybody around me is dying — when’s it going to be my turn?”
The difficulties of these relationships affect parents as well as their children. Judy Longfellow’s son was in the hospital at one time with another boy close to Mark’s age. The other boy, says Judy, “left the hospital and he died the night he left. It was the feeling of everybody — everybody knew — that he was going home to die. That really bothered Mark. I think from that time on he really began to think about his disease a little more than he might have had that not happened. And he was good buddies with another little boy who died too.” Yet she made no attempt to shield her son from such occurrences. “He liked those people; he had a good time with them. And even though it ended up to be a sad moment in his life, I don’t think you can protect people from life and expect them to be well-rounded individuals, you just can’t do it. Life is life, you know, you take the good and the bad.”
The advisability of having CF patients associate with one another is an issue which has been widely debated within the CF medical community. The position of the San Diego CF Center is quite clear, however. An annual camp for CF kids, periodic meetings at the CF House for adolescents, adults, and CF parents, along with the fostering of a recognizable “CF family” in San Diego, are evidence of Dr. Harwood’s belief that as a physician he has “no role in protecting people from knowledge.… There are some aspects of this disease that patients know a hell of a lot more about than I do because they live with the disease every day. So if a family — or children, which they frequently are — chooses to get involved with another patient with this disease who may be severely involved or even dying, that is their business. And I don’t feel any need to prevent it from happening. In fact I would say people with the disease teach each other an awful lot about how to live with the disease, or even how to die with the disease, that that be something they want to know about.”
They have taught Dr. Harwood, too, about dying with the disease. His experience with CF has led him to adopt the role of advisor rather than prescriber when someone is near death, with the patient and family making the ethical and moral decisions which must be made at that time — such as whether to cease or continue giving medical care, and whether to die at home or in the hospital (with the home being favored by nearly 80 percent of the center’s patients). His evolving attitude about the role of the physician at the time of death has been forced on him, he says, by a couple of patients in his past “who were smart enough and brave enough to tell me and the other caregivers here what it’s really all about, when it’s clear that my science isn’t going to extend a person’s life beyond a certain point.” So-called “extraordinary” medical measures and hospitalization up until the very moment of death are rarely practiced, says Harwood, because “our patients have told us very clearly: there’s a limit. There is a limit and we’re defining it, not you the physician but me, the patient.”
One such patient was a Carlsbad woman named Rita Bowers, who died last September at the age of 35. “She had been in the hospital in May,” said her husband, Rick. “She was in there for three weeks and it was a very bad time for her. It was the worst time she had ever spent in there, and when she left they weren’t optimistic at all about her chances. I talked to Ivan about it and he said, ‘If she keeps going the way she is, probably two weeks.’ And Rita said, ‘No, I’m not ready to die. I’ve still got too much to do. I’m leaving the hospital and I’m going home…that’s where I’m going to die, where I can be with the people I love.’ ”
Her doctors visited her at home throughout the summer, while family members changed intravenous medicines, gave treatments, and virtually duplicated the medical care she would have received in the hospital — until the last week. “The last week,” Rick recalls, “she refused to take anything — no medications, no IVs, no treatments, no anything. And that really hurt — hurt me. I felt it hard, because of all the years and trouble we’d gone to. For so long we’d had to do treatments three times a day, the meds and everything else, and when she stopped that last week, it really hurt.”
Rita’s last months were spent preparing; preparing her two children, spending time with them, talking with them — “I’m going to be leaving you, I don’t know when. But even though I’m not here physically anymore, I’ll be with you all of your life”; ordering the details of her life; and preparing herself. When she died, says Rick, “she said she was ready.”
Being ready to die, being prepared, is extremely important, says Bob Bourquin. “Dying before you’re ready is one of the toughest ways to go, it’s one of the most painful. When the kids are in the hospital and they’re going to fight it and they’ve gotta keep going, they’re frantic and they’re dying, that’s what’s so hard to see, because you just know there’s nothing you can say or do, they are just so full of anxiety and fear. If people have died and they were somewhat willing to accept death, it’s not as hard for us who are left.”
The Cystic Fibrosis Foundation calls CF a “hidden handicap” — you can’t readily see that it affects a person the way you can with, say, the loss of a limb. But the label refers also to the way the disease can gnaw at a person’s spirit as well as his health. One CF adult who found himself unable to work after having done so for many years commented, “You can go for long periods of time and forget that you have a serious illness. And then suddenly you get sick and you get very depressed and think, ‘Oh, I’m sick again, here I have to face this.’ And it’s always going to be that way. It’s a gradual deterioration and you have to keep adjusting to that deterioration — that’s what’s so difficult to do; you have to keep lowering your expectations, lowering your degree of involvement in activities and living less and less. Trying to live as fully as you can but being able to do less and less all the time.”
Yet for at least one San Diego couple this development has created unsuspected possibilities. Michael and Lisa (not their real names) are in their mid-30s, live in La Jolla, and have a daughter, in spite of Michael’s having CF (the vast majority of CF males being sterile). Michael is tanned, has a good physical build, and looks as if he spends most of his time on a tennis court when not in his office. In short, he looks the successful Southern California professional, which he is. But after a three-week hospital stay in December and years of thinking about it “ad nauseam,” he recently decided to quit his job.
As his health had worsened over the years, the strain of managing both disease and career had proven increasingly difficult. His employers showed something of a chronic inability to understand this, even after their trim young executive’s “allergies” and “colds” were explained to them, and when he informed them he was quitting, they asked, “What is it? Has there been some change in your health?”
There had, following CF’s inexorable downhill path, but Michael’s attitude had changed, too. Upon returning to work after the December hospitalization, he had realized that he “just didn’t have a great interest in picking up the accumulated piles of stuff, clearing them off, and charging onward and upward... I asked myself, 'What good does it do me to make this kind of money?'
"We went on a vacation to Yellowstone for two weeks last summer, and the day before we left I started feeling lousy and having some Vipmontvsis. For the next six days, either in the middle of the night or next morning, I'd get up and cough up a bucket of blood. I said, 'By God, I'm going to enjoy this vacation,' so we continued on and got to everywhere we were goign to go, but it was not like we had planned. I had these great visions of taking my daughter horseback riding, and we were going river rafting, and we were going to take hikes in the woods. We took one hike in the woods and I started coughing up blood. And so I asked, 'What good is all this money?'
"I had fought as long as I felt I could fight. I was just tired of fighting. I had always put myself in a position of saying the work comes first; if I got sick and needed a tune-up, I'd say, 'No, I can't afford to take two weeks off So everything else suffered. I'd go home at night and collapse, and weekends I'd lie around collapsed and kind of recuperate for Monday morning, when I'd go back into the office and drag through the week. I'd go to the office feeling lousy and I'd go on trips feeling lousy. It was a real problem as far as doing anything other than the duties from nine to five. Sometimes, if I was real sick, I'd go home at lunch for an extra treatment."
Difficulties developed at work because his associates could not comprehend the narrowing limitations of this child's disease with the innocuous symptoms — coughing and fatigue. He who was once the favored, fair-haired boy, solver of tough problems, was now "dogging it or wasn't motivated."
"I'd always been success and achievement oriented," savs Michael, "and I had to resolve whether I was a quitter. Was I a failure? But I've come to the point where I don't want to be looking back a year form now, or five years from now, or whatever, and saying I wasted the last year or five years. I want to feel I've gotten something out of life, done something I want to do other than grind out an earning.... It's an opportunity for a new beginning."
"Most people could put up with a shitty job," says his wife, "and say, 'I'm going to retire in 30 years.' Well, that's out for him, for us. So you have to refocus your life, you have to do something different, find new values." And in a peculiar sort of paradox, reaching the point of this decision has removed a great deal of the burden of uncertainty as to how CF will affect their lives. "We are at this point looking ahead," says Lisa, "we really are. For the first time we are looking ahead and I feel good about it."
Few people Michael and Lisa associate with seem able to understand their decision. The comments they have met with—"He doesn't seem that sick," "What job are you going to have Monday morning?" "How are you going to bring in the money?" — seem to underscore an almost irreconcilable schism between a person with a concrete perception that his life is finite, and a society vitally concerned, as Lisa says, with how people "are going to arrange their houses and what room they're going to add on and whether the maid should come every Tuesday or Thursday." The person with CF can't help but see things differently.
Not only is work viewed differently—as more than a means of "grinding out an earning"—but, according to Bob Bourquin, so are social relationships. He says the person with a shortened life span subjects the ordinary social niceties to greater scrutiny. "If, for example, I feel like a friendship is unsatisfactory, I will totally write it off, and other people can't understand that. I think it partly comes from the fact that people with CF have a feeling that time is more precious, more fleeting, and why spend your time with somebody that you don't really like, thatyou don't really have something with? I think there is more of a tendency just to walk away from people like that, and I don't think that's bad. I don't believe in maintaining a friendship just for the sake of maintaining a friendship. It sounds trite, but the quality of time that you spend with someone is so much more important that the quantity."
The feeling that time is fleeting, that experience must be maximized, work made valuable, and life lived from day to day comes in virtually every conversation with a member of the CF community. There is an awareness that a shortened life span requires not holding back but giving more, and it comes through both in Rick Bowers's comment that Rita's outlook was, let's cut through all the crap and get to the meat," and in Judy Longfellow's saying, "We never avoided doing something with Mark because he had cystic fibrosis. We took Mark places a lot of times where I know he really didn't feel good, he maybe shouldn't have been there, he would be tired or something, but he wanted to go."
The young father of a CF baby recently expressed a wish to take his son camping and fishing. "I'd like him to play baseball. I'd like him to do whatever he wants to do,because I'm going to enjoy him, we're going to enjoy each other." But a moment later he exposed the dilemma of CF by adding, "I'm a realist. We want things to be as normal as they can, but there might be — there will be—some unpleasant things occurring." His wife, though, had no doubts on how to face this future. "A full life," she said, "can be three years, six years, whatever we have. But we have to work as hard as we can to provide him with what we are able to give him so he can live a good, full life to the best of his ability and of our ability."
The mother of a child dead many years confirmed that this is the only workable approach. "There is a great deal of satisfaction," she said, "when you feel that a child has been happy and had the best of care and you've done everything that you could do."
Cystic fibrosis, like many diseases, affects the mind as much as it does the body. The ever-present cough and the accompanying sputum serve as continual reminders of the temporal and fragile qualities of life. The pool of one's own blood hypnotically fixes the gaze after a hemoptysis, and its presence has a peculiarly powerful ability to make other things—tomorrow's appointment with a client, the upcoming calculus final — miraculously dwindle in significance.
Every night the person with CF runs through the regimen, reviews the litany: "Did I take all my pills today? The enzymes before every meal and snack, the antibiotics, bronchodilators, vitamins; the profusion of gelatinous shapes and colors. Did I take them all? Did I get enough exercise? Did I do the treatments that were necessary? Was everything loosened and expelled that was there to be loosened and expelled?" And unavoidably the question arises: "Does it matter? In the morning it will all be there again."
It does matter. And tomorrow you'll do it all again. You'll wonder again, "Am I as healthy today as I was the day before? And will I be this healthy again tomorrow?" And you'll worry again, "Am I doing everything, everything that can be done?" Not to beat it, because you can't, but at least to hold your own. But much as it demands and takes, cystic fibrosis also gives. Throughout it all — amongst the survivors, the families, the "victims" themselves — inextricably intertwined with the sadness, the grief, and the anguished projections into the future, in among all these threads of sorrow runs also one of deep beauty. It comes from facing with dignity what has been dictated by fate, from persevering in the face of encroaching debilitation; from knowing what the end will bring, yet living each day, and the day after, and the day after that, as if each were of vintage quality; it comes from a gleeful anticipation of each day's newly opened blossoms and from the song of the mockingbird that wakes you in the night. It comes from the satisfaction in learning what is and what is not important. And it comes from fighting the best you can.
Here's something you might be interested in.
San Diego cystic fibrosis sufferers learn to live life by the moment
No time to dream
San Diego cystic fibrosis sufferers learn to live life by the moment
No time to dream
Intensive chest therapy involves physically beating on the patient’s chest.
"When I was nine my grandmother heard from a friend about this clinic down in Mississippi on the Gulf Coast. So my mother and I flew down there in the winter because I was constantly up and down, sick some days, well the next — missed a lot of school. We went down to this miracle clinic where they gave you this elixir with arsenic in it; stayed there a week, must have been a very expensive thing. We came back and I started taking the miracle elixir; did that a few months and my parents noticed I was going bald — I was losing all my hair. So they stopped the elixir and miraculously my hair started growing back... I don't think it changed things much, the miracle elixir didn't do a whole lot, but it tasted horrible, I do remember that. It was pink and it tasted like the inside of somebody's tennis shoe. When I had to take that during the day it was like, oh no!! But I took it because I thought I was going to get better; none of us knew about CF. We were ignorant. We were babes in the woods." — Bob Bourquin
Bob Bourquin. When Bob walks in, is 26, looks like a football player, those parents have got to feel some resentment.
Ellen Derner is 29 and works in a southeast San Diego kindergarten as a teacher’s aide. Her father shied away from the miracle elixirs, but 20 years ago, when she too was nine, he quit his job, sold everything that wouldn’t fit in the back of a U-Haul, loaded Ellen, her mother, and two sisters in the family car, left his native Montreal, and started driving south — to somewhere it didn’t snow. Ellen had spent the previous summer with an aunt in Florida, where the series of colds and the terrible, persistent cough which had prompted the desperate move seemed to have subsided. Florida was too humid though, so they drove west. When they reached Arizona, Ellen stopped coughing, but her mother didn’t want to live in Arizona and they continued on to Los Angeles. The smog in L.A. was unbearable so they again turned south, coming to a stop in San Diego.
Ellen Derner: “As it turns out, the climate doesn’t make any difference."
“As it turns out, though,” says Ellen, “the climate doesn’t make any difference, and about a year after we got here I got sick again. It was diagnosed [falsely] as tuberculosis and I was bedridden for eight or nine months…they were pretty sure I was going to die. My parents regretted we’d left Canada then.” But they stayed, suffering through the unexplained illnesses and colds until a firm diagnosis was made when she was 19.
Mark Longfellow.was eating his mother out of house and home — 18 pancakes at one sitting.
At that time she went to University Hospital, where a test was performed in which a weak electrical current and a special chemical were applied to her arm, causing a small area of skin to sweat. After careful collection and measurement, it was determined that her sweat contained an abnormally high amount of salt; the test was positive. However, her insurance plan required that she go to another hospital for treatment, and there she was retested three times, each time with a negative result. However, she was somewhat dubious about their technique. “They put my whole body in a plastic bag with just my head sticking out the top and waited for me to sweat so they could collect and measure it.” But it could have been worse, according to Ellen. “They used to wrap kids in wool blankets and make them run around the hospital.”
Rita Bowers: "No, I’m not ready to die. I’ve still got too much to do."
She herself never doubted the outcome of the tests. “I knew I had all the symptoms. So I finally went back to University and paid to have it redone.” Positive again. She had cystic fibrosis.
The disease first appeared in the scientific literature in 1936 as congenital cystic pancreatic fibromatosis and bronchiectasis syndrome, a mouthful of a name that basically meant doctors were finding some very sick babies with a puzzling combination of symptoms previously thought unrelated: chronic lung infection in patients who also developed fibrous and cystlike scarring of the pancreas.
Dr. Ivan Harwood “can’t tell anybody they’re going to get better."
By 1938 researchers were satisfied that the strange combination was caused by a single disease and they trimmed the name to fibrocystic disease of the pancreas, but still not a lot was known about it. Doctors were telling mothers, “We don’t know why it is, but these babies all follow the same pattern. You can’t find any food to agree with them, and then they get pneumonia and die.” When the accepted name became cystic fibrosis a few years later, the disease had become distinguished, said one textbook, “for the frequency with which it was correctly diagnosed for the first time at post mortem.”
CF patients cannot properly expel the thick secretions they produce and the mucus remains trapped in the lungs.
A more useful method of diagnosis was developed as a result of observations made during a New York City heat wave in 1948. As is common during a heat wave, hospital emergency rooms began filling up with victims of heat stroke brought on by excessive salt and fluid loss. A pair of doctors noted that a curiously high number of these persons seemed to have cystic fibrosis and they began wondering why. It was soon discovered that the sweat of these patients contained up to ten times the normal amount of salt — explaining the high incidence of heat stroke and also forming the basis for a diagnostic test.
Yet it is unfortunate that the sweat test Ellen Derner described is so often incorrectly done, because it is the only conclusive test for an incurable and poorly understood disease. Cystic fibrosis (CF) occurs in one-fourth of the offspring from two parents who carry the genetic trait; it is the greatest genetic killer of young people in this country, with as many as 1 out of every 20 Caucasians believed to carry the gene. Such carriers are not affected by the disease, however, and since they show absolutely no symptoms, cannot be identified; despite the highly promising work of several researchers, at the moment the only sure way of identifying a carrier of CF is by working backward: the parents of a child with CF must carry the gene.
The basic genetic defect which is responsible for cystic fibrosis remains unknown, but it is known that this defect causes excessively thick and sticky secretions of mucus. This thick mucus brings on an almost unbelievable variety of problems, the most serious being chronic and progressive lung infections, and inefficient digestion resulting from clogged digestive ducts. Mucus is produced by the lungs as their primary means of fighting infection (which is why people get congested when they have a cold), but CF patients cannot properly expel the thick secretions they produce and the mucus remains trapped in the lungs, where it fosters the growth of bacteria — which in turn promotes the secretion of more mucus, which then provides a home for more bacteria. It is this circular pattern of lung infections which eventually proves fatal.
In the ’50s a CF child could not be expected to reach school age. By the mid-’60s the average life expectancy had grown to age 11. Now, with better treatment and earlier detection, the national Cystic Fibrosis Foundation projects that half of all CF patients born today will live past the age of 21. But increased longevity has in many cases become something of a mixed blessing for longer-lived patients who have become grown-ups burdened with a disease perceived almost exclusively — both by the general medical community and the public — as an affliction of children only.
The executive director of the Cystic Fibrosis Foundation’s local chapter is Winnie Burke, an energetic and outspoken woman in her mid-40s who directs the chapter’s fund-raising and public-education activities from her office on Fourth Avenue in Hillcrest. “When I first took this job,” she says, “the first time I got close to a kid who died, I sat here in my office and I cried and cried. The other woman who was working in the office at that time came in and she said, ‘If you’re going to react like that every time one of these kids dies, I don’t think you’re going to be suited for this job.’ I looked at her and I said, ‘If I don’t react this way, I don’t think I’m going to be suited for this job.’ ” In the intervening ten years, Burke has had no shortage of opportunities to adjudge her job fitness, since this series of events repeats itself in her office with distressing regularity.
Like virtually anyone whose life has been touched by cystic fibrosis, Winnie Burke learned its lessons quickly. Doctors have called it “the great masquerader.” It can be subtle, whimsical, merciless, and above all, arbitrary. For a CF patient, life is a misshapen path of sharp turns and detours about which nothing is predictable except that it always leads downhill, sometimes gently, sometimes steeply. The disease is maddeningly erratic; what happens to one person is no indication of what will happen to another. Within a single family one child might die at a few years of age while another makes it into his 30s to become one of CF’s “old folks.” But such reprieves are understood to be only temporary. Today’s apparent good health, the brisk seven-mile run, the 75-mile bike ride, the workout at the gym, all too frequently give way to next week’s ten-day hospital stay. One minute’s laughter becomes the next minute’s wrenching cough, which, sometimes, leads to the burst artery and coughed-up blood of hemoptysis. And if it doesn’t, there is always the fear.
If these are extremes — the more common story of CF being one of frail, barrel-chested children, of frequently missed school and canceled family vacations — they are not atypical; it is a disease which defies neat description. It is a disease Camus or Sartre would have understood, for it teaches the meaning of existentialism as few things can. It is a disease which causes five- and six-year-olds to think deeply about sorrow and joy, the unfairness of fate, and why people are born to die.
In 1967 cystic fibrosis became part of the life of an attractive, composed Coronado woman named Judy Longfellow. Her son, Mark, was 14 months old at the time and she recognized, she says, “that something was wrong. He had diarrhea an awful lot and was eating me out of house and home — 18 pancakes at one sitting, that sort of thing; it was incredible. It was some time before we came up with the diagnosis, because after we had tried eliminating different foods [to check for allergies], we did a sweat test at Mercy Hospital which came back negative. But our doctor was very suspicious that Mark had CF, so we took him up to Children’s Hospital for more work. They did several tests and they all came back positive.
“There was a panel of doctors, there were like three, and we went into this room — Ralph [her husband] wasn’t with me. It was a very dark dismal kind of room, a room that would hold maybe 30 people, and we sat in this little nucleus and they basically told me Mark had CF, and they didn’t really elaborate much at all except to say that it was a very harsh disease. Then they sent me on my way.”
But a few days later Mrs. Longfellow and her husband were provided with a few details by their specialist at Children’s.
“He came into the room and he said, ‘Well, you might as well know right now that if your son coughs, that’s just about the end; he will probably not last three months. He’ll never be able to go to school.’ The picture he painted for us was very, very black. So we were left with absolutely no hope that this child would live to be two years old.… His manner was very matter-of-fact, he was in the room maybe three minutes; he went through all this stuff and then he was gone. That angered me considerably.”
Mark first exhibited the deep, thick cough of CF when he got a lung infection about six months after this. The doctor’s three-month time limit proved wrong, however, and Mark did go to school — where he excelled. He was tested at the age of six and found capable of doing college-level math. “He loved math,” said his mother, “just loved it.” He was also an avid baseball player and one year he became a Little League all-star. Not until last spring, when Mark was 14 years old, did he die.
Mark was a child with a lot of friends, and in spite of his disease he led an active life, but it was a life punctuated by visits to the hospital. In contrast to some CF kids, who by the age of 13 may have been in as many as 30 times, Mark, says his mother, “wasn’t in really a lot. Maybe seven times. There was one period where he went for almost two years without a hospital stay.”
Usually such hospitalizations are for what is referred to in CF jargon as a “tune-up.” Tune-ups become necessary when a cold stirs up the bacteria in a CF patient’s lungs; they usually last 10 to 14 days, and their main treatments are the administration of intravenous drugs and intensive chest physical therapy that involves physically beating on the patient’s chest. This can be done with either a mechanical vibrator or by hand in a clapping manner which evokes the sound of a horse’s clippity-clop; a few minutes spent observing this procedure can yield a keen perspective on the limitations of modern medicine. The purpose of this activity is to loosen and dislodge the thick, sticky mucus (termed “junk”) which a person with CF can cough up in mind-boggling quantities after a treatment. One adult commented that he coughed up eight ounces a day when he felt well and twice that when he had an infection.
CF patients are advised to do treatments at home as well as in the hospital; they can be done by a child’s parents, or, when old enough to use the special vibrators, children can do their own. The Longfellows came to view the treatments as a part of life, a habit “like brushing your teeth every morning. It took an hour every time we did them and we routinely did them twice a day.” But while some patients have no doubt about the value of treatments (one proclaiming, for instance, that “if I stopped doing my treatments today I would probably be in the hospital in two weeks, maybe not even that long”), others avoid them fanatically because they symbolize the unremitting grip CF maintains upon those it affects: two, three, sometimes up to four hours a day spent clearing one’s lungs in a ceaseless effort simply to stay alive. One mother said, “It’s just like pulling hen’s teeth” to get her 11-year-old son to take a treatment. “He hates it. He thinks if he works on himself he doesn’t need that pounding. In the morning he goes in and he coughs and he coughs and he gets most of that stuff up himself. He’ll cough for maybe five or ten minutes.”
It is one of CF’s more unnecessary tragedies that the type of callous (and often ignorant) welcome to the medical world which Judy Longfellow received is far from uncommon. And though families have had the amazing luck of having a child correctly diagnosed the very first time he got sick, the stories of wrong diagnoses, missed diagnoses, and refused diagnoses are legion among the CF community. One man told of having “pneumonia, or some problem, as an infant”; “repeated illnesses” all through his youth; “what was diagnosed as viral pneumonia three times” in his early teens; “mononucleosis during college”; “and other problems off and on.” One physician was “convinced” he had tuberculosis and “kept running skin tests. He couldn’t understand why they kept turning out negative.” He spoke of being given 40 different diagnoses — with any one as good as another — and of being treated for “allergies” until his mid-20s.
It was the ironic ill fortune of this man, now 35, to be too healthy and longlived to be considered a candidate for cystic fibrosis. Nor is this sort of episode confined to the backwoods of medicine: as recently as two years ago, and based upon the erroneous results of a sweat test conducted by placing a plastic baggie over one hand, a pulmonary specialist at one of San Diego’s most respected medical institutions persisted in treating for tuberculosis an adult patient who had been clearly diagnosed elsewhere as having CF — presumably because the patient was too old and too well to be “cystic.”
Just as common as the litany of false diagnoses and incorrect treatments are the attendant tales of mental turmoil compounded by physicians who failed either to recognize the medical subtleties and variability of CF or to comprehend and appreciate the emotional difficulties generated by the disease, or both. When Ellen Derner was finally diagnosed, she was understandably relieved, as she put it, to “find out what it was, to learn that the symptoms were all related and that it had a name.” But her doctors, she says, couldn’t understand that. “One of them stood in my room and yelled at me that this was something that was going to kill me and that I had better start taking it seriously. They couldn’t understand what a relief it was to have it all explained, to have it all make sense.”
The medical mistakes are understandable given that virtually nothing in CF is orderly — diabetes, liver disease, and sterility can develop in addition to the respiratory and digestive disorders; and it can be found among blacks and Hispanics, though it is disproportionately a disease of whites. But what is harder to understand is that the predominant attitude within the medical community seems based on an image of CF drawn largely from the ’50s. The general medical community maintains a picture of CF similar to that put forth recently by the prestigious New England Journal of Medicine. In an editorial, they pronounced CF “a grim sentence, usually prolonged through an unhappy adolescence to a sad, inevitable end.” This attitude, unfortunately, is often reflected in the medical care dispensed by physicians who hold it, and it is this situation which contributes to the unrestrained joy most CF patients and families feel when they finally do find a physician familiar with their disease.
The San Diego Cystic Fibrosis and Pediatric Pulmonary Disease Center at University Hospital is 1 of 125 specialized centers supported by the Cystic Fibrosis Foundation. The medical care of approximately 160 local CF patients is supervised here by Dr. Ivan Harwood, center director, and Dr. Nancy Olmsted, center codirector.
The center’s administrative facilities and offices are located two blocks south of the hospital in a former residence on Front Street that has since been named “CF House.” One of these offices is used by Dr. Harwood; its appearance tells a great deal about the man who, says one of his patients, “can’t tell anybody they’re going to get better. They can plateau and remain stable, but all of his patients are going to die.” On the walls hang photographs of children he has cared for; in a corner sits an old wooden desk littered with papers and a wide assortment of items: a microscope which appears older than van Leeuwenhoek’s; a small ceramic elephant; a rack of smoking pipes; and the disordered components of a unique filing system referred to by others in the office as the CFIS — the Cystic Fibrosis Information Sink, into which correspondence and other printed matter often vanish for wondrously long periods of time.
Ivan Harwood came to San Diego in 1970 and joined the faculty at UCSD a year later. He came at a time when about half a dozen patients were being seen twice monthly at a clinic established through the efforts of the local CF chapter. He didn’t come to San Diego intending to run the CF Center. “I didn’t plan it,” he says. “I sort of fell into this backward. There were a few people who needed care and there was a need that wasn’t being met.” There was also another appeal, something Harwood refers to as “the chemistry between a few patients and a doctor.”
Few physicians are attracted to the care of the chronically ill. It subverts their image as healers; to say it can be disheartening is an understatement. One member of the hospital CF team estimates that they lose roughly one patient every six weeks. This often leads doctors to immerse themselves in the scientific aspects of medicine, depersonalizing the patient and the disease. Avoiding this form of self-protection has required Dr. Harwood to develop a perspective which takes into account the limitations of specializing in a disease without a cure. “If I approached CF as a disease where I always lost or where there was nothing to be done,” he says, “it would be so dismal and hopeless that I couldn’t continue to be involved with it very long. So I think what I’ve done is to say well, okay, my role as a physician is not necessarily just to cure a disease — because I can’t cure this one, I accept that from the start — but I can do an awful lot at some stages of the disease to prolong life, and I can do certain things in the end stages to make a very difficult part of one’s journey through life a little better, a little easier. So I’ve gotten to the point where I say, well, that’s part of the whole business. It’s difficult, I accept that, but I also have the satisfaction of being competent with patients in that very difficult stage — patients that usually, because of the nature of the disease, we have gotten to know very well and to care about.”
The San Diego CF Center utilizes a “team approach” in caring for its patients. In one sense the team is the group of professionals responsible for ministering medical care in all its forms — the doctors, nurses, technicians, therapists, and social workers. But as an outgrowth of Dr. Harwood’s belief that “the ultimate responsibility for somebody’s care is in themselves or their parents,” patients and families are also strongly encouraged to take part in decisions concerning their medical care. “It really works better for us to make the family and the patient partners in the whole process of trying to manage a disease that can’t be cured.”
A dual perspective on CF and its care is offered by one respiratory therapist at University Hospital. Bob Bourquin has CF in addition to being a member of the medical team. He is 26 (and still has his hair despite his subjection to the “miracle elixir cure”); he says there are some absolutes in CF “and there’s an absolute with me: I’m going to live another five years. It’s not definite but I think that’s reasonable.”
Growing up in Maryland, he spent a sickly childhood with “asthma” and “allergies” until the day he “turned purple” when he was nine. “I couldn’t breathe at all. It was just like I was shut down.” He recovered after a prolonged hospitalization during which doctors made the diagnosis of CF and told his parents, “Your child has cystic fibrosis and he’s going to die.”
He spent a lot of time after that trying to deny he had CF. “That’s pretty much how I approached the thing when I was in my teens. Even when I was really sick I’d never do treatments. I could be so sick I could climb a flight of steps and almost black out — in school the 50-yard dash was more like the 50-yard crawl — and still I wouldn’t do anything for it.
“I knew so little about CF six or seven years ago. So, so, very little. And you have all these preconceived notions which you get from the newspaper articles. Like somewhere I’d read the average life span of somebody with CF was 21. I figured I was 19; I had two years to live. I thought that was it — everybody died when they were 21. I didn’t realize at the time, because I was totally uneducated medically, that some people live a lot longer, some people live a lot shorter. I really thought that when I turned 21 that would be like the last supper. I’d cut the birthday cake and I’d go in and lie down. And die.”
When he was 19 an incident occurred which forced him to realize what CF would mean: he spent five weeks in the hospital as a result of air leakage from his lungs into his chest cavity — a pneumothorax. “That was the big turning point. I could not deny it anymore. I started doing treatments every day. I spent the entire fall of that year like a self-imposed cripple, paranoid of having to go back in the hospital. I was extremely depressed, did nothing except lie around the house in utter paranoia and despair.”
Shortly after this Bourquin began obtaining medical care from a CF center in Cleveland. His spirits started to improve as he met other CF patients and grew to realize that he could take a direct hand in his medical care. “It’s not sheer happenstance that I’m where I am now. I work hard at it. And it is hard to get in an hour and a half, because that’s how long my treatment takes me every day. It’s hard to do that and still go to work and do everything else.”
Remembering the reassurance that can come from meeting other, especially older, CF patients, Bourquin makes it a point to spend part of his time at University Hospital talking with the parents of CF children. It is a task he has learned to approach with delicacy. “If the children are fairly healthy, or even moderately healthy, the parents are universally pleased, relieved, and delighted to see me there,” he says. “They can look at me and say, ‘Wow, that’s neat, maybe my kid is going to grow up.’ If, on the other hand, their kid’s really sick — when one of the CF kids is dying and I’m the therapist — that’s when I really get the resentment vibes, or the jealousy vibes. You know, when Bob walks in, is 26, looks like a football player, I feel somewhat guilty. Those parents have got to feel some resentment; at that point they know their kid’s not going to make it and they can’t help but be jealous that I have. That’s just basic human nature.
“There’s a whole lot of vibrations between CF patients. It’s very awkward when you’re healthier. Vibrations go out to you of jealousy and anger — I’ve been the recipient of those. When you see somebody who’s sicker than you it’s tough because your heart really goes out to them. You know yourself when you’ve been sick how you feel, and when you see someone with CF who’s dying, who’s close to death, it’s hard to look at them because you know what it’s like and you know what it’s going to be like; you just sort of are instinctive about it. On the other hand, it’s the old animal instinct: thank God it isn’t me.”
Other reactions can also mark the relationships formed between people with CF: comfort and relief can come through learning that others share the same plight, the same experiences, the same worries; despondency can result from watching friends fall like so many dominoes, from thinking, “Here are X number of young adults and Monica died, and so-and-so died, everybody around me is dying — when’s it going to be my turn?”
The difficulties of these relationships affect parents as well as their children. Judy Longfellow’s son was in the hospital at one time with another boy close to Mark’s age. The other boy, says Judy, “left the hospital and he died the night he left. It was the feeling of everybody — everybody knew — that he was going home to die. That really bothered Mark. I think from that time on he really began to think about his disease a little more than he might have had that not happened. And he was good buddies with another little boy who died too.” Yet she made no attempt to shield her son from such occurrences. “He liked those people; he had a good time with them. And even though it ended up to be a sad moment in his life, I don’t think you can protect people from life and expect them to be well-rounded individuals, you just can’t do it. Life is life, you know, you take the good and the bad.”
The advisability of having CF patients associate with one another is an issue which has been widely debated within the CF medical community. The position of the San Diego CF Center is quite clear, however. An annual camp for CF kids, periodic meetings at the CF House for adolescents, adults, and CF parents, along with the fostering of a recognizable “CF family” in San Diego, are evidence of Dr. Harwood’s belief that as a physician he has “no role in protecting people from knowledge.… There are some aspects of this disease that patients know a hell of a lot more about than I do because they live with the disease every day. So if a family — or children, which they frequently are — chooses to get involved with another patient with this disease who may be severely involved or even dying, that is their business. And I don’t feel any need to prevent it from happening. In fact I would say people with the disease teach each other an awful lot about how to live with the disease, or even how to die with the disease, that that be something they want to know about.”
They have taught Dr. Harwood, too, about dying with the disease. His experience with CF has led him to adopt the role of advisor rather than prescriber when someone is near death, with the patient and family making the ethical and moral decisions which must be made at that time — such as whether to cease or continue giving medical care, and whether to die at home or in the hospital (with the home being favored by nearly 80 percent of the center’s patients). His evolving attitude about the role of the physician at the time of death has been forced on him, he says, by a couple of patients in his past “who were smart enough and brave enough to tell me and the other caregivers here what it’s really all about, when it’s clear that my science isn’t going to extend a person’s life beyond a certain point.” So-called “extraordinary” medical measures and hospitalization up until the very moment of death are rarely practiced, says Harwood, because “our patients have told us very clearly: there’s a limit. There is a limit and we’re defining it, not you the physician but me, the patient.”
One such patient was a Carlsbad woman named Rita Bowers, who died last September at the age of 35. “She had been in the hospital in May,” said her husband, Rick. “She was in there for three weeks and it was a very bad time for her. It was the worst time she had ever spent in there, and when she left they weren’t optimistic at all about her chances. I talked to Ivan about it and he said, ‘If she keeps going the way she is, probably two weeks.’ And Rita said, ‘No, I’m not ready to die. I’ve still got too much to do. I’m leaving the hospital and I’m going home…that’s where I’m going to die, where I can be with the people I love.’ ”
Her doctors visited her at home throughout the summer, while family members changed intravenous medicines, gave treatments, and virtually duplicated the medical care she would have received in the hospital — until the last week. “The last week,” Rick recalls, “she refused to take anything — no medications, no IVs, no treatments, no anything. And that really hurt — hurt me. I felt it hard, because of all the years and trouble we’d gone to. For so long we’d had to do treatments three times a day, the meds and everything else, and when she stopped that last week, it really hurt.”
Rita’s last months were spent preparing; preparing her two children, spending time with them, talking with them — “I’m going to be leaving you, I don’t know when. But even though I’m not here physically anymore, I’ll be with you all of your life”; ordering the details of her life; and preparing herself. When she died, says Rick, “she said she was ready.”
Being ready to die, being prepared, is extremely important, says Bob Bourquin. “Dying before you’re ready is one of the toughest ways to go, it’s one of the most painful. When the kids are in the hospital and they’re going to fight it and they’ve gotta keep going, they’re frantic and they’re dying, that’s what’s so hard to see, because you just know there’s nothing you can say or do, they are just so full of anxiety and fear. If people have died and they were somewhat willing to accept death, it’s not as hard for us who are left.”
The Cystic Fibrosis Foundation calls CF a “hidden handicap” — you can’t readily see that it affects a person the way you can with, say, the loss of a limb. But the label refers also to the way the disease can gnaw at a person’s spirit as well as his health. One CF adult who found himself unable to work after having done so for many years commented, “You can go for long periods of time and forget that you have a serious illness. And then suddenly you get sick and you get very depressed and think, ‘Oh, I’m sick again, here I have to face this.’ And it’s always going to be that way. It’s a gradual deterioration and you have to keep adjusting to that deterioration — that’s what’s so difficult to do; you have to keep lowering your expectations, lowering your degree of involvement in activities and living less and less. Trying to live as fully as you can but being able to do less and less all the time.”
Yet for at least one San Diego couple this development has created unsuspected possibilities. Michael and Lisa (not their real names) are in their mid-30s, live in La Jolla, and have a daughter, in spite of Michael’s having CF (the vast majority of CF males being sterile). Michael is tanned, has a good physical build, and looks as if he spends most of his time on a tennis court when not in his office. In short, he looks the successful Southern California professional, which he is. But after a three-week hospital stay in December and years of thinking about it “ad nauseam,” he recently decided to quit his job.
As his health had worsened over the years, the strain of managing both disease and career had proven increasingly difficult. His employers showed something of a chronic inability to understand this, even after their trim young executive’s “allergies” and “colds” were explained to them, and when he informed them he was quitting, they asked, “What is it? Has there been some change in your health?”
There had, following CF’s inexorable downhill path, but Michael’s attitude had changed, too. Upon returning to work after the December hospitalization, he had realized that he “just didn’t have a great interest in picking up the accumulated piles of stuff, clearing them off, and charging onward and upward... I asked myself, 'What good does it do me to make this kind of money?'
"We went on a vacation to Yellowstone for two weeks last summer, and the day before we left I started feeling lousy and having some Vipmontvsis. For the next six days, either in the middle of the night or next morning, I'd get up and cough up a bucket of blood. I said, 'By God, I'm going to enjoy this vacation,' so we continued on and got to everywhere we were goign to go, but it was not like we had planned. I had these great visions of taking my daughter horseback riding, and we were going river rafting, and we were going to take hikes in the woods. We took one hike in the woods and I started coughing up blood. And so I asked, 'What good is all this money?'
"I had fought as long as I felt I could fight. I was just tired of fighting. I had always put myself in a position of saying the work comes first; if I got sick and needed a tune-up, I'd say, 'No, I can't afford to take two weeks off So everything else suffered. I'd go home at night and collapse, and weekends I'd lie around collapsed and kind of recuperate for Monday morning, when I'd go back into the office and drag through the week. I'd go to the office feeling lousy and I'd go on trips feeling lousy. It was a real problem as far as doing anything other than the duties from nine to five. Sometimes, if I was real sick, I'd go home at lunch for an extra treatment."
Difficulties developed at work because his associates could not comprehend the narrowing limitations of this child's disease with the innocuous symptoms — coughing and fatigue. He who was once the favored, fair-haired boy, solver of tough problems, was now "dogging it or wasn't motivated."
"I'd always been success and achievement oriented," savs Michael, "and I had to resolve whether I was a quitter. Was I a failure? But I've come to the point where I don't want to be looking back a year form now, or five years from now, or whatever, and saying I wasted the last year or five years. I want to feel I've gotten something out of life, done something I want to do other than grind out an earning.... It's an opportunity for a new beginning."
"Most people could put up with a shitty job," says his wife, "and say, 'I'm going to retire in 30 years.' Well, that's out for him, for us. So you have to refocus your life, you have to do something different, find new values." And in a peculiar sort of paradox, reaching the point of this decision has removed a great deal of the burden of uncertainty as to how CF will affect their lives. "We are at this point looking ahead," says Lisa, "we really are. For the first time we are looking ahead and I feel good about it."
Few people Michael and Lisa associate with seem able to understand their decision. The comments they have met with—"He doesn't seem that sick," "What job are you going to have Monday morning?" "How are you going to bring in the money?" — seem to underscore an almost irreconcilable schism between a person with a concrete perception that his life is finite, and a society vitally concerned, as Lisa says, with how people "are going to arrange their houses and what room they're going to add on and whether the maid should come every Tuesday or Thursday." The person with CF can't help but see things differently.
Not only is work viewed differently—as more than a means of "grinding out an earning"—but, according to Bob Bourquin, so are social relationships. He says the person with a shortened life span subjects the ordinary social niceties to greater scrutiny. "If, for example, I feel like a friendship is unsatisfactory, I will totally write it off, and other people can't understand that. I think it partly comes from the fact that people with CF have a feeling that time is more precious, more fleeting, and why spend your time with somebody that you don't really like, thatyou don't really have something with? I think there is more of a tendency just to walk away from people like that, and I don't think that's bad. I don't believe in maintaining a friendship just for the sake of maintaining a friendship. It sounds trite, but the quality of time that you spend with someone is so much more important that the quantity."
The feeling that time is fleeting, that experience must be maximized, work made valuable, and life lived from day to day comes in virtually every conversation with a member of the CF community. There is an awareness that a shortened life span requires not holding back but giving more, and it comes through both in Rick Bowers's comment that Rita's outlook was, let's cut through all the crap and get to the meat," and in Judy Longfellow's saying, "We never avoided doing something with Mark because he had cystic fibrosis. We took Mark places a lot of times where I know he really didn't feel good, he maybe shouldn't have been there, he would be tired or something, but he wanted to go."
The young father of a CF baby recently expressed a wish to take his son camping and fishing. "I'd like him to play baseball. I'd like him to do whatever he wants to do,because I'm going to enjoy him, we're going to enjoy each other." But a moment later he exposed the dilemma of CF by adding, "I'm a realist. We want things to be as normal as they can, but there might be — there will be—some unpleasant things occurring." His wife, though, had no doubts on how to face this future. "A full life," she said, "can be three years, six years, whatever we have. But we have to work as hard as we can to provide him with what we are able to give him so he can live a good, full life to the best of his ability and of our ability."
The mother of a child dead many years confirmed that this is the only workable approach. "There is a great deal of satisfaction," she said, "when you feel that a child has been happy and had the best of care and you've done everything that you could do."
Cystic fibrosis, like many diseases, affects the mind as much as it does the body. The ever-present cough and the accompanying sputum serve as continual reminders of the temporal and fragile qualities of life. The pool of one's own blood hypnotically fixes the gaze after a hemoptysis, and its presence has a peculiarly powerful ability to make other things—tomorrow's appointment with a client, the upcoming calculus final — miraculously dwindle in significance.
Every night the person with CF runs through the regimen, reviews the litany: "Did I take all my pills today? The enzymes before every meal and snack, the antibiotics, bronchodilators, vitamins; the profusion of gelatinous shapes and colors. Did I take them all? Did I get enough exercise? Did I do the treatments that were necessary? Was everything loosened and expelled that was there to be loosened and expelled?" And unavoidably the question arises: "Does it matter? In the morning it will all be there again."
It does matter. And tomorrow you'll do it all again. You'll wonder again, "Am I as healthy today as I was the day before? And will I be this healthy again tomorrow?" And you'll worry again, "Am I doing everything, everything that can be done?" Not to beat it, because you can't, but at least to hold your own. But much as it demands and takes, cystic fibrosis also gives. Throughout it all — amongst the survivors, the families, the "victims" themselves — inextricably intertwined with the sadness, the grief, and the anguished projections into the future, in among all these threads of sorrow runs also one of deep beauty. It comes from facing with dignity what has been dictated by fate, from persevering in the face of encroaching debilitation; from knowing what the end will bring, yet living each day, and the day after, and the day after that, as if each were of vintage quality; it comes from a gleeful anticipation of each day's newly opened blossoms and from the song of the mockingbird that wakes you in the night. It comes from the satisfaction in learning what is and what is not important. And it comes from fighting the best you can.
Comments